
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Exploiting senescence as a therapeutic target in osteoarthritis
Christopher Larbie1, Umar Raza2
1Department of Biochemistry and Biotechnology, Kwame Nkrumah University of Science and Technology (KNUST), Kumasi, Ghana.
2School of Basic Medical Sciences, Shenzhen University, Shenzhen, Guangdong Province, China.
Correspondence: Christopher Larbie (Department of Biochemistry and Biotechnology, Kwame Nkrumah University of Science and Technology (KNUST), Kumasi, Ghana; E-mail: clarbie.cos@knust.edu.gh).
Asia-Pacific Journal of Pharmacotherapy & Toxicology 2024, 4: 85-94. https://doi.org/10.32948/ajpt.2024.11.19
Received: 25 Sep 2024 | Accepted: 17 Nov 2024 | Published online: 22 Nov 2024
Key words osteoarthritis, senescence, therapeutic target, senescence-associated secretory phenotype
Regenerative therapies for OA are riskier in older patients, contrasting with focal cartilage defects where phenotypic stability of chondrocytes is crucial for success [18]. A senescent microenvironment may also hinder stem cell implantation for joint repair [19]. Therefore, understanding and exploring senescence-related complexities may offer potential therapeutic insights for OA and related conditions. In this context, various treatment approaches are emerging to target cellular senescence in OA. First and foremost are senolytic drugs that induce apoptosis and clear senescent cells by countering anti-apoptotic pathways. On the other hand, senomorphic drugs that modulate the SASP are being actively investigated for their role in OA treatment [22]. In addition, stem cell rejuvenation is also being explored as a tool to circumvent senescence in OA [20]. In this review, we discuss the detrimental impacts of senescence in OA and how therapeutic options targeted towards senescence are changing the landscape of management and treatment options for OA.
Chondrocyte senescence
Senescence in chondrocytes, the key cells responsible for maintaining cartilage integrity, and other joint tissues, contribute to significant clinical outcomes in OA patients [7]. While chondrocytes are typically hypo-replicative during homeostasis, they may proliferate in specific contexts. For instance, chondrocytes form 'clusters' as an apparent effort to repair damaged matrix during the early stages of OA [21]. In this scenario, the interplay between quiescence and senescence is complex, as mitogenic stimulation of damaged, quiescent cells can trigger the induction of senescence they re-enter the cell cycle [22]. Unfortunately, the prevalence of senescent chondrocytes along with acquisition of SASP increases with age, serving as a pivotal contributor to the initiation and progression of OA [23]. One of the primary clinical observations related to chondrocyte senescence is the progressive damage to the cartilage matrix. Senescent chondrocytes exhibit reduced capacity for ECM synthesis and repair, leading to the gradual degradation of cartilage [24]. This degradation is clinically evident through joint pain, stiffness, and reduced joint function, as the loss of cartilage integrity compromises joint stability and mobility. Patients often report difficulty in performing daily activities and experience limitations in range of motion due to chondrocyte-related cartilage damage [25]. Moreover, senescence-related changes in chondrocytes contribute to alterations in the ECM composition marked by increased production of degenerative enzymes like MMPs [26]. These changes are clinically visible through imaging modalities such as X-rays and MRIs, which show joint space narrowing, osteophyte formation, and structural deformities indicative of cartilage damage and joint degeneration [27]. Additionally, extracellular vesicles (EVs) secreted by OA-related senescent chondrocytes induce senescence in neighboring non-senescent cells and suppress chondrogenesis through a para-senescent effect [28]. In summary, altered ECM composition, inflammatory responses, and acquisition of SASP are key events linking chondrocyte senescence to disease progression and clinical manifestations in OA.
Synovial senescence
Senescence in synovial fibroblasts, along with synovial macrophages, osteoblasts, and adipocytes, contributes to the production of SASP and acquisition of inflammatory environment [29, 30], that plays a crucial role in cartilage degeneration, subchondral bone remodeling, and ultimately leads to cartilage loss, OA development and progression [31, 32]. Synovial tissue contributes significantly to the senescence burden in OA knee joints [12], with SASP levels markedly upregulated in isolated synovial tissue from human OA patients, exacerbating synovial inflammation [33]. Senescence can disrupt the synovial microenvironment and drive OA-like degeneration, as demonstrated by mouse knee joints transplanted with senescent fibroblasts which exhibited cartilage erosion, osteophyte formation, and loss of mobility [34]. Synovial cells are responsible for producing approximately 55% of cytokines, and interactions between synoviocytes and chondrocytes are vital in the pathogenesis of OA [35]. These interactions can lead to the degradation of ECM in articular cartilage, which subsequently exacerbates synovial inflammation [36]. This inflammation promotes MMP13 production, and resulting build-up of matrix debris aggravates synovial inflammation, creating a vicious cycle that ultimately results in total joint destruction. Key proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 are pivotal in driving synovial inflammation and cartilage destruction [37]. Elevated levels of cytokines such as IL-6 in the synovial fluid of OA patients indicate their potential involvement in triggering senescence and SASP in surrounding cells [38]. These cytokines also promote the formation of SASP in senescent chondrocytes, with IL-1β notably enhancing the production of other proinflammatory cytokines and MMPs in chondrocytes [39]. Similarly, TNF-α induces the expression of MMPs and ADAMTS, contributing to further degradation of the cartilage matrix [40]. Cytokines, released in SASP, can also, in turn, upregulate MMPs, such as MMP13 and ADAMTS-5, which contribute to ECM degradation in cartilage [41]. This cartilage degeneration, along with synovitis and joint inflammation, leads to clinical symptoms such as joint swelling, and tenderness observed in OA patients [42]. In summary, senescence in synovial fibroblasts and associated release of SASP profoundly aid in disease progression in OA.
Stem cell senescence
In addition to senescent chondrocytes and synovial cells, age and/or stress related senescence in MSCs play crucial role in overproducing SASP factors such as chemokines, cytokines, and MMPs, resulting in detrimental effects on cell responses to growth factors and their proliferative capacities [43]. One notable marker of senescent MSCs is the elevated expression of p16INK4a, a potent cell cycle inhibitor. Studies using animal models have shown that p16INK4a-positive senescent MSCs, particularly those residing in the articular osteochondral region, can contribute significantly to cartilage damage and the progression of OA [44]. Within the synovial tissue of OA joints, the p16INK4a protein regulates the production of SASP factors, which have a catabolic effect on tissues [45]. Furthermore, analyses of synovial fluid from OA patients have revealed elevated levels of SASP factors like IL-6 [46]. Prolonged exposure to IL-6 in synovial fluid derived MSCs hinders their differentiation into chondrocytes [47], a critical process for cartilage regeneration. Similarly, IL-6 inhibits the differentiation of murine bone marrow-derived MSCs into chondrocytes, highlighting its dose-dependent regulatory role in cartilage maintenance [48]. Articular cartilage repair and rejuvenation rely heavily on the functional capacity of chondrocytes and resident MSCs, particularly synovial MSCs, which possess superior chondrogenic potential compared to other MSC populations [49]. Their ability to produce hyaline cartilage matrices is crucial for cartilage regeneration. However, an excessive presence of senescent synovial MSCs can impede these regenerative processes. Notably, senescent synovial MSCs are resistant to apoptosis and exhibit prolonged survival. Their accumulation in joints, cartilage, synovial tissue, and synovial fluid is a common observation in various pathological conditions [50]. Although inflammatory environments can activate and promote the proliferation of synovial MSCs, OA-related persistent inflammation can deplete these cells and induce replicative senescence. A significant population of senescent synovial MSCs resides in OA synovium, characterized by features such as cellular senescence, heightened production of pro-inflammatory molecules, and diminished chondrogenic potential [51]. Despite all this, the precise molecular mechanisms that resident senescent synovial MSCs employ to disrupt homeostasis and impair cartilage function remain a subject of ongoing research and exploration.
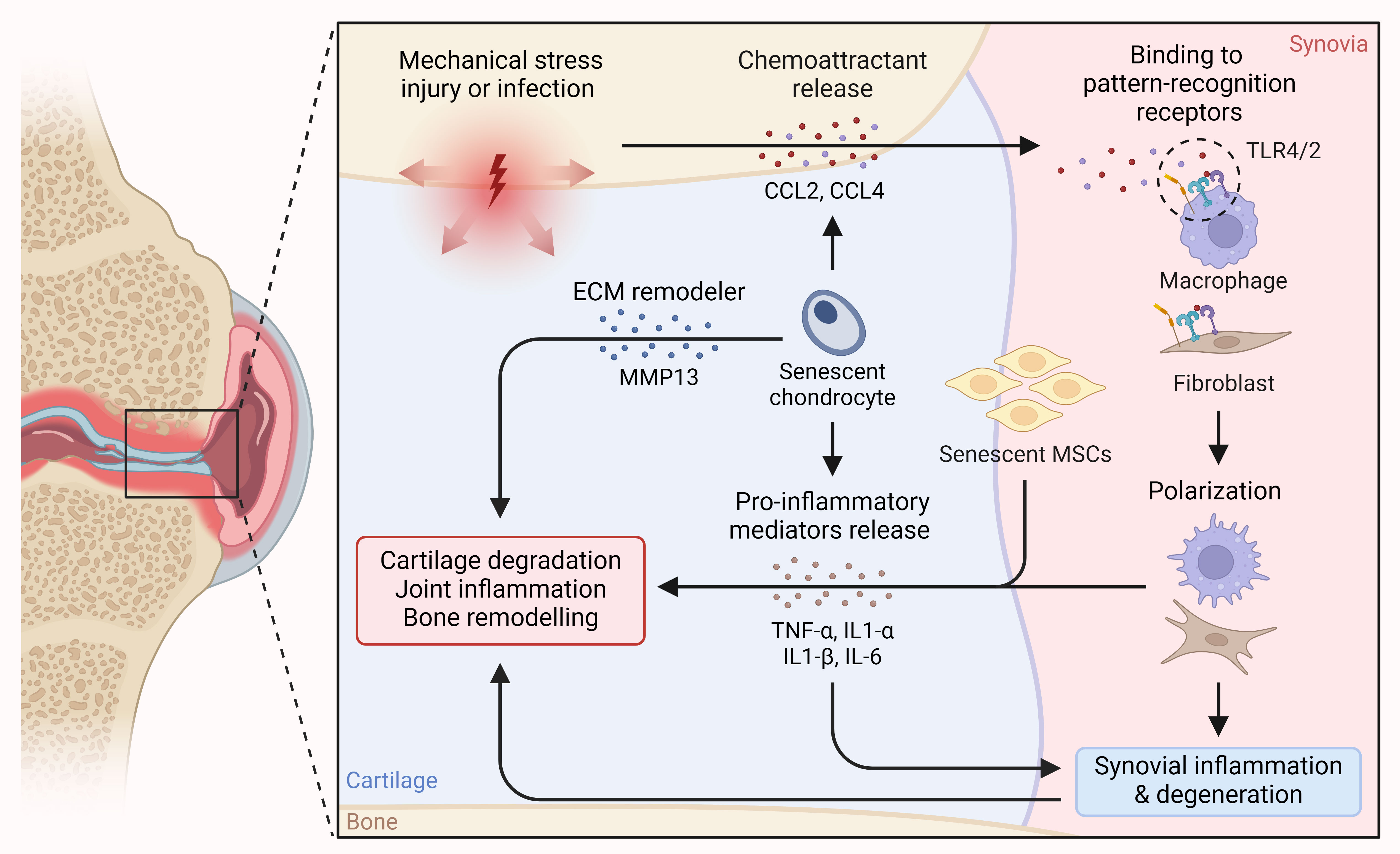 Figure 1. Detrimental role of senescence in OA. Chemoattractants released in response to mechanical stress and from senescent chondrocytes in OA lead to immune cell polarization and infiltration into cartilage, resulting in the release of pro-inflammatory cytokines. Simultaneously, the release of pro-inflammatory mediators from senescent chondrocytes and MSCs exacerbates the situation, ultimately leading to cartilage and synovial degeneration, joint and synovial inflammation, and bone remodeling.
Figure 1. Detrimental role of senescence in OA. Chemoattractants released in response to mechanical stress and from senescent chondrocytes in OA lead to immune cell polarization and infiltration into cartilage, resulting in the release of pro-inflammatory cytokines. Simultaneously, the release of pro-inflammatory mediators from senescent chondrocytes and MSCs exacerbates the situation, ultimately leading to cartilage and synovial degeneration, joint and synovial inflammation, and bone remodeling.
Senolytics – Killing senescence
In a groundbreaking preclinical investigation, a novel inducible transgene was engineered to selectively eliminate senescent cells expressing elevated levels of p16 [53]. Mice carrying this transgene exhibited an extended median lifespan and delayed onset of age-related pathologies compared to their wild-type counterparts. Notably, there was a substantial reduction in the development of post-traumatic OA, when a similar transgenic approach was employed to clear senescent cells specifically in mouse articular cartilage [12]. While these experiments primarily utilized transgenic mice to induce apoptosis in senescent cells, other studies explored whether senolytics could replicate these effects therapeutically. For instance, a study investigating gene expression profiles of senescent versus proliferating cells revealed upregulation of genes associated with anti-apoptotic signaling networks during senescence, including BCL-2 family members and proteins in the PI3K–AKT pathway [54]. Many senolytics selectively trigger apoptosis in senescent cells by inhibiting pro-survival pathways that are activated in senescent cells but not in healthy ones. Notably, navitoclax (ABT-263), a dual inhibitor of BCL-2 and BCL-XL, administered to irradiated or naturally aged mice, depleted senescent haematopoietic stem cells and muscle stem cells, fostering cellular rejuvenation [55]. Navitoclax reduced the senescence burden in cartilage explants by inducing apoptosis in p16-expressing chondrocytes, effectively eliminating them. [56]. Another example is the senolytic combination of dasatinib and quercetin, currently under investigation in clinical trials for treating idiopathic pulmonary fibrosis associated with senescence [57, 58]. Dasatinib inhibits various tyrosine kinases, while quercetin, a plant flavonol, inhibits PI3K and serine proteinase inhibitors (serpins) [59]. Treatment with dasatinib and quercetin in mice attenuated the detrimental effects of senescence, improving healthspan and lifespan by reducing senescent osteocytes in bone and enhancing bone microarchitecture [60]. While these drugs await human trials for joint tissue diseases, several senolytics, including UBX0101, are currently under investigation for OA treatment [23]. UBX0101 inhibits the interaction between p53 and mouse double minute 2 homologue (MDM2), selectively clearing senescent cells in post-traumatic OA, decreasing proteoglycan loss, and alleviating OA-related symptoms [12].
Treatment with SIRT-activating compounds exhibit chondroprotective effects in OA. Resveratrol, a natural anti-oxidant and a potent SIRT1 inducer, has been shown to inhibit MMP-13 expression in human OA chondrocytes through SIRT1 activation [61]. Additionally, it protects against articular cartilage destruction in OA mice by suppressing the NF-κB and HIF-2α pathways in a SIRT1-dependent manner [62]. Studies in preclinical models have highlighted resveratrol's ability to reduce serum levels of inflammatory cytokines such as IL-1β, IL-6, and TNF-α, alleviating pain and stiffness and potentially improving knee joint function in OA patients, with favorable safety and tolerability profiles [63]. Metformin, an anti-diabetic medication, has recently been shown to suppress the expression of p16, IL-6, and MMP-13 while promoting the expression of Collagen type II and Aggrecan in OA chondrocytes by regulating the miR-34a/SIRT1 axis [64]. Vitamin D deficiency accelerates age-related knee OA development, characterized by cartilage surface destruction, proteoglycan loss, and chondrocyte senescence with SASP acquisition. Supplementation with vitamin D3 reverses OA phenotypes rescuing chondrocyte proliferation, matrix protein synthesis, and reducing oxidative stress via upregulating SIRT1 expression [65]. Since an elevated serum ratio of SIRT1 N-terminal to its C-terminal fragments correlates with early-stage OA and chondrosenescence, the concomitant administration of systemic navitoclax and intra-articular UBX0101 reduced the ratio by clearing senescent cells [66, 67]. One notable synthetic SIRT-activating compound, SRT1720, a robust SIRT1 activator, has demonstrated significant reduction in MMP-13 and ADAMTS-5 expression in chondrocytes, mitigating articular cartilage degeneration and osteophyte formation while attenuating synovial inflammation, thus delaying OA progression in murine models [68]. Similarly, SRT2104, a selective SIRT1 activator, exhibits efficacy in preventing knee OA progression in mice by lowering MMP-13, ADAMTS-5, IL-1β, IL-6 levels, acetylated NF-κB p65, and enhancing Col-II synthesis within cartilage [69]. Its systemic benefits include anti-inflammatory and antioxidant effects linked to reduced NF-κB activity, mirroring the physiological improvements seen with SRT1720 [70]. Notably, non-therapeutic clinical studies confirm the excellent safety profile and tolerability of SRT2104 [71]. Cyanidin, an activator of SIRT6, increases SIRT6 expression in a dose-dependent manner. It significantly reduces IL-1β-induced expression of pro-inflammatory mediators like TNF-α, IL-6, COX-2, MMP-13, and ADAMTS-5, thereby mitigating synovial inflammation and cartilage degradation. In OA mouse models, cyanidin downregulates MMP-13 expression and upregulates Col-II expression, preserving articular cartilage matrix integrity [72]. Chondrocytes from younger donors repair damage more efficiently than those from older donors. Activation of SIRT6 with MDL-800 improved repair, reduced baseline DNA damage in older chondrocytes and aging murine cartilage, indicating a potential role in mitigating DNA damage and delaying senescence, which could impact OA progression [73]. Flavonoids like fisetin, known for activating SIRTs and linked to longevity, has demonstrated efficacy in inhibiting IL-1β-induced inflammation in osteoarthritic chondrocytes, mitigating cartilage erosion, ECM degradation, apoptosis, and senescence in OA. These compounds are currently undergoing clinical trials for OA symptom alleviation [74-76].
The exploration of high-throughput drug screening for senolytics targeting chondrocytes and synovial cells, along with the identification of novel mechanisms contributing to OA pathology, has gained significance. In a specific study, over 1,000 compounds were screened for senolytic activity in a human chondrocyte cell line, revealing that fenofibrate, a flavonoid and peroxisome proliferator-activated receptor-α (PPARα) agonist used to treat dyslipidaemias, induced apoptosis in SA-β-gal-positive chondrocytes [77]. This finding prompted an investigation into reduced PPARα expression in the blood and knee cartilage of OA patients.
Concerns regarding the use of senolytics in OA treatment arise from potential side effects and variations in drug potency. The impact of promoting cell death on tissue integrity, exacerbating cartilage and bone loss in OA patients, remains uncertain. Notably, a study in mice using diphtheria toxin on superficial zone proteoglycan-expressing cells did not induce further cartilage damage but rather improved injury outcomes [78]. This challenges assumptions about the consequences of promoting cell death in OA treatment and suggests a potential benefit in preventing injury-induced cartilage loss by eliminating senescent chondrocytes. Another consideration in employing senolytics for OA is the conflicting evidence regarding the role of senescence in physiological processes. While senescence is often implicated as a driver of aging and disease, studies suggest a beneficial role in tissue remodeling and wound healing [79]. For instance, senescent cells accumulated during limb regeneration in salamanders but were naturally cleared before full regrowth. Efficient immunosurveillance of senescent cells might play a crucial role in tissue repair, as demonstrated in studies involving macrophage-mediated clearance and secretion of growth factors by senescent cells near wound sites [80, 81]. Consequently, further research is essential to determine if wholesale elimination of senescent cells from joints could lead to side effects contributing to tissue loss in OA.
Senomorphics – modulating SASP
The strategic targeting of pathways and molecules associated with inflammation and disease has long been a therapeutic approach. A diverse range of senomorphic candidates has emerged, demonstrating the ability to suppress SASP-related mechanisms without inducing apoptosis [23]. These include inhibitors of IκB kinase and NFκB, such as NEMO-binding domain peptides [82], JAK inhibitors like ruxolitinib [83], ATM inhibitors exemplified by KU-60019 [84], compounds disrupting progerin–lamin A/C binding, for instance, JH4 [85], activators of PDGF and fibroblast growth factor signaling [86], TGFβ receptor type 2 and p21 inhibitors [87]. The link between SASP factor expression and OA highlights the potential of inhibiting these factors in OA. However, selecting appropriate target is crucial to guarantee therapeutic efficacy and specificity.
Despite the increased interest in exploring cytokine inhibitors and neutralizing antibodies approved by the FDA for inflammatory conditions like OA, In vivo experiments using IL-1 blockade and IL-1 knockout in mice did not show a reduction in cartilage destruction or synovial inflammation in collagenase-induced OA [88]. Nevertheless, such inhibitors present intriguing prospects for OA therapies due to their established safety and efficacy in different hereditary autoinflammatory conditions [89], though pre-clinical and clinical studies targeting cytokines for OA treatment have yielded mixed results. For instance, the combined blockade of IL-1α and IL-1β using monoclonal antibodies prevented the development of pain and reversed established pain in a preclinical model of OA [90]. Alternatively, Lutikizumab, a dual inhibitor of IL-1α and IL-1β, showed limited pain improvement and no impact on synovitis in a phase II trial for knee OA patients [91]. Similarly, etanercept, a TNF inhibitor, failed to alleviate pain and had minimal effects on structure in a trial for inflammatory hand OA [92]. Canakinumab, a selective IL-1β neutralizing antibody currently undergoing phase 2 clinical trials for knee OA patients (NCT04864392), has shown significant reductions in serum C-reactive protein (CRP) levels and a lower risk of cardiovascular events in patients with a history of myocardial infarction and elevated baseline CRP. These findings were observed in the large Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) [93]. Notably, a post-hoc analysis of the CANTOS study suggested that participants randomized to canakinumab exhibited lesser OA symptoms and underwent fewer OA-related joint replacements compared to the placebo group [94]. IL-6 is a potential target for OA therapy, and systemic administration of an IL-6 neutralizing antibody has been shown to decrease cartilage lesions and subchondral bone sclerosis in experimental OA model in vivo [95]. Additionally, Systemic administration of the anti-IL-6-receptor neutralizing antibody (MR16) resulted in the improvement of cartilage damage, reduction in synovial inflammation, and inhibition of osteophyte formation in vivo [96]. Despite this, IL6 knockout mice exhibit more severe OA in response to aging than wild-type mice, suggesting the complexity of OA pathogenesis requires a multifaceted treatment approach [97]. In line with this, tocilizumab, an IL-6 neutralizing antibody approved for RA treatment, did not show benefits in a phase 3 clinical trial for hand OA, as it indicated no improvement in pain or secondary outcomes, while slightly more frequent adverse events were observed [98]. IL-17 significantly contribute to the pathogenesis of OA and is strongly linked to joint pain in OA patients [99]. In addition, IL-17 producing Th17 cells are known to induce senescence in fibroblasts. Underscoring the roles of these cells and IL-17 in OA development and cartilage senescence, intraarticular injection of an IL-17-neutralizing antibody has been shown to alleviate cartilage degeneration and reduce the senescence marker p21 in vivo [15]. However, the effectiveness of IL-17 neutralizing antibodies in human OA remains to be tested. Given their catabolic impact on cartilage, MMPs represent an additional category of SASP factors that should be considered as promising therapeutic targets. Specifically, MMP13 stands out as the highly expressed MMP in connective tissue [100] and serves as the key enzyme for the degradation of type-II collagen [101]. Chondrocytes from individuals with OA were observed to exhibit elevated MMP13 levels compared to chondrocytes from those with healthy cartilage [102]. Moreover, transgenic mice overexpressing MMP13 develops OA-like arthropathy, suggesting MMP13's pivotal role in OA pathogenesis [103]. A separate study demonstrated that the chondrocyte-specific deletion of MMP13 reduces OA severity. In addition, treating wild-type mice with a selective MMP13 inhibitor, CL82198, reduces OA severity, increases type II collagen levels, and inhibit chondrocyte death [104].
Exogenous supplementation with itaconate, an anti-inflammatory metabolite activates Nrf2, inhibits the STING-dependent NF-κB pathway, and reduces SASP-driven inflammation, ECM degeneration, and chondrocyte senescence. Itaconate also regulate macrophage polarization, decreasing chondrocyte apoptosis [105]. Supplementation with naturally occurring falvonoid, procyanidin B2, dampens IL-1β-triggered SASP factors expression, preserving the extracellular matrix (ECM), reducing chondrocyte apoptosis, and attenuating senescence via repressing Nrf2/NF-κB pathway. Additionally, procyanidin B2 exhibited anti-apoptotic properties through the Nrf2/BAX/Bcl-2 pathway and alleviated knee cartilage degeneration in an OA rat model [106]. Pinitol, is a natural compound that counteracts the effects of TNF-α-induced cellular senescence and cell cycle arrest by rescuing NRF2 signaling in chondrocytes, thus alleviating OA [107]. Piperlongumine, an amide alkaloid constituent of long pepper, reduces senescence markers and SASP, preserves cartilage matrix components, mitigates oxidative stress and DNA damage, and decreases inflammatory markers associated with OA [108]. Dendrobine, an alkaloid found in orchids, effectively combats OA by inhibiting ECM degradation and SASP factors expression, improving mitochondrial function, and suppressing NF-κB activation in chondrocytes [109]. Similarly, supplementation with antioxidant, pyrroloquinoline quinone, prevents articular surface collapse, reduces cartilage matrix protein loss, and inhibits oxidative stress, DNA damage, cellular senescence, and inflammatory cytokine secretion associated with OA development, highlighting its potential as a preventive measure against OA progression [110]. Collectively, these findings suggest that inhibiting SASP factors through senomorphics holds promise for OA treatment. However, further research is essential to identify specific SASP factors contributing to OA pathology and determine if their inhibition can effectively slow or prevent disease progression.
Rejuvenation of stem cells to prevent senescence in OA
Increasing age and pathological factors alters the extracellular environment, impacting the fate of all cells within a tissue, including resident MSCs. Understanding the mechanisms involved could aid in stem cell rejuvenation, maintaining MSC function and preserving healthy tissues in older age and disease scenarios [111]. Local administration of exosomes derived from bone marrow-MSCs of young rats significantly accelerates bone regeneration and improves mechanical properties in older rats, affirming the potential of stem cell rejuvenation in rejuvenating aged and senescent tissues [112]. In the context of OA, bone marrow derived-MSCs primed with TNF-α and IFN-γ have exhibit increased anti-inflammatory potential to reduce SASP-driven inflammation in a chemically induced OA model [113]. Notably, yes-associated protein (YAP) represses MSC senescence via promoting FOXD1 transcription. Intra-articular delivery of YAP or FOXD1 coding lentiviral particles has been shown to decrease senescent cells, while also mitigating articular inflammation and cartilage erosion. These findings suggest that gene therapy which introduces geroprotective factors to rejuvenate senescent MSCs could represent a new tool for OA therapy in the future [114]. Recently, antler stem cells (ASCs)-derived exosomes (ASCs-EVs) have demonstrated remarkable potential to induce proliferative and regenerative capacity in human senescent MSCs, thereby rejuvenating these cells. Following treatment with ASCs-EVs, human MSCs showed reduced signs of senescence, and intra-articular injection of ASCs-EVs alleviated cartilage damage in an OA mouse model [20]. These results suggest that ASCs could serve as a sustainable source of EVs for cell-free therapy. Further explorations are warranted to fully tailor stem cell rejuvenation as a tool to prevent senescence in OA.
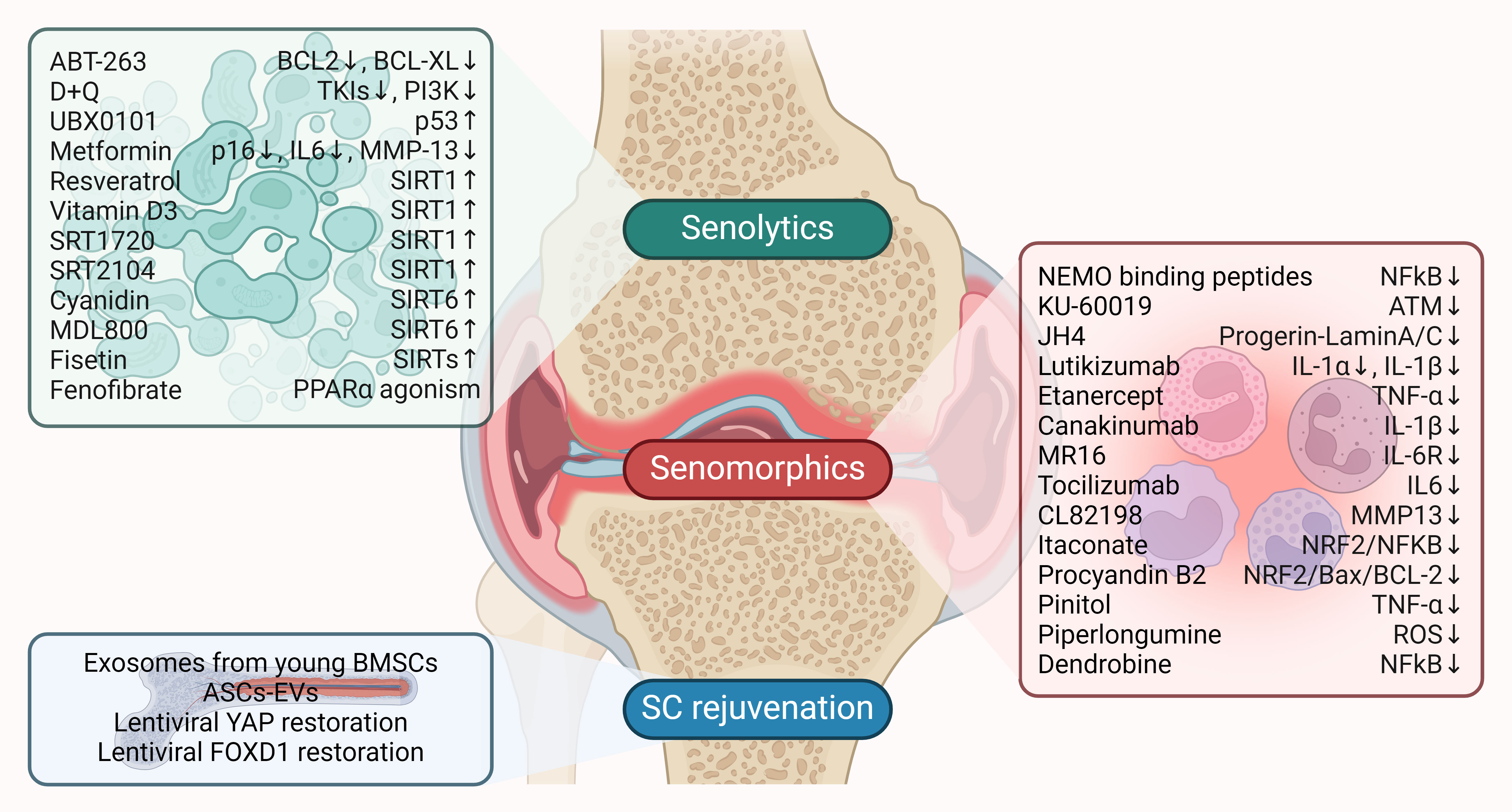 Figure 2. Therapeutic targeting of senescence in OA. Senolytics, senomorphics, and stem cell (SC) rejuvenation are key therapeutic options currently being investigated to target senescence in OA. Senolytic and senomorphic drugs are listed along with their impact on molecular targets. Arrows indicate the effect on the target: ↑ (upregulation) and ↓ (downregulation).
Figure 2. Therapeutic targeting of senescence in OA. Senolytics, senomorphics, and stem cell (SC) rejuvenation are key therapeutic options currently being investigated to target senescence in OA. Senolytic and senomorphic drugs are listed along with their impact on molecular targets. Arrows indicate the effect on the target: ↑ (upregulation) and ↓ (downregulation).
No applicable.
Ethics approval
No applicable.
Data availability
The data will be available upon request.
Funding
None.
Authors’ contribution
CL contributed to draft, critical revision of the article and approved the final manuscript; UR was devoted to to figure production and modification.
Competing interests
None.
- Dillon CF, Rasch EK, Gu Q, Hirsch R: Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol 2006, 33(11): 2271-2279.
- Zhang Y, Jordan JM: Epidemiology of osteoarthritis. Clin Geriatr Med 2010, 26(3): 355-369.
- Buckwalter JA, Saltzman C, Brown T: The impact of osteoarthritis: implications for research. Clin Orthop Relat Res 2004, https://doi.org/10.1097/01.blo.0000143938.30681.9d. Epub ahead of print.(427 Suppl): S6-15.
- Honkonen SE: Degenerative arthritis after tibial plateau fractures. J Orthop Trauma 1995, 9(4): 273-277.
- Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R et al: Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 2016, 7: 11190.
- Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W: Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016, 22(1): 78-83.
- Zhang XX, He SH, Liang X, Li W, Li TF, Li DF: Aging, Cell Senescence, the Pathogenesis and Targeted Therapies of Osteoarthritis. Front Pharmacol 2021, 12: 728100.
- Childs BG, Zhang C, Shuja F, Sturmlechner I, Trewartha S, Fierro Velasco R, Baker D, Li H, van Deursen JM: Senescent cells suppress innate smooth muscle cell repair functions in atherosclerosis. Nat Aging 2021, 1(8):698-714.
- Diekman BO, Sessions GA: Expression of p16(INK)(4a) is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 2018, 17(4): e12771.
- Del Rey MJ, Valín Á, Usategui A, Ergueta S, Martín E, Municio C, Cañete JD, Blanco FJ, Criado G, Pablos JL: Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun Ageing 2019, 16: 29.
- Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, Drake MT, Tchkonia T, LeBrasseur NK, Kirkland JL et al: Identification of Senescent Cells in the Bone Microenvironment. J Bone Miner Res 2016, 31(11): 1920-1929.
- Jeon OH, Kim C, Laberge RM, Demaria M: Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 2017, 23(6): 775-781.
- Martin JA, Brown T, Heiner A, Buckwalter JA: Post-traumatic osteoarthritis: the role of accelerated chondrocyte senescence. Biorheology 2004, 41(3-4): 479-491.
- Schwab W, Schulze-Tanzil G, Mobasheri A, Dressler J, Kotzsch M, Shakibaei M: Interleukin-1beta-induced expression of the urokinase-type plasminogen activator receptor and its co-localization with MMPs in human articular chondrocytes. Histol Histopathol 2004, 19(1): 105-112.
- Faust HJ, Zhang H, Han J, Wolf MT, Jeon OH, Sadtler K, Peña AN, Chung L, Maestas DR, Jr., Tam AJ et al: IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J Clin Invest 2020, 130(10): 5493-5507.
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M: Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr Biol 2017, 27(17): 2652-2660.e2654.
- Vinatier C, Domínguez E, Guicheux J, Caramés B: Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front Physiol 2018, 9: 706.
- Saris DB, Vanlauwe J, Victor J, Haspl M, Bohnsack M, Fortems Y, Vandekerckhove B, Almqvist KF, Claes T, Handelberg F et al: Characterized chondrocyte implantation results in better structural repair when treating symptomatic cartilage defects of the knee in a randomized controlled trial versus microfracture. Am J Sports Med 2008, 36(2): 235-246.
- Li J, Hansen KC, Zhang Y, Dong C, Dinu CZ, Dzieciatkowska M, Pei M: Rejuvenation of chondrogenic potential in a young stem cell microenvironment. Biomaterials 2014, 35(2): 642-653.
- Lei J, Jiang X, Li W, Ren J: Exosomes from antler stem cells alleviate mesenchymal stem cell senescence and osteoarthritis. Protein Cell 2022, 13(3): 220-226.
- Lotz MK, Otsuki S, Grogan SP, Sah R, Terkeltaub R, D'Lima D: Cartilage cell clusters. Arthritis Rheum 2010, 62(8): 2206-2218.
- Ogrodnik M, Salmonowicz H, Jurk D, Passos JF: Expansion and Cell-Cycle Arrest: Common Denominators of Cellular Senescence. Trends Biochem Sci 2019, 44(12): 996-1008.
- Coryell PR, Diekman BO, Loeser RF: Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol 2021, 17(1): 47-57.
- Rim YA, Nam Y, Ju JH: The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int J Mol Sci 2020, 21(7): 2358.
- He Y, Li Z: Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models. Biology (Basel) 2020, 9(8): 194.
- Maldonado M, Nam J: The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed Res Int 2013, 2013: 284873.
- Newman S, Ahmed H, Rehmatullah N: Radiographic vs. MRI vs. arthroscopic assessment and grading of knee osteoarthritis - are we using appropriate imaging? J Exp Orthop 2022, 9(1): 2.
- Jeon OH, Wilson DR, Clement CC, Rathod S, Cherry C, Powell B, Lee Z, Khalil AM, Green JJ, Campisi J et al: Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 2019, 4(7): e125019.
- Jeon OH, David N, Campisi J, Elisseeff JH: Senescent cells and osteoarthritis: a painful connection. J Clin Invest 2018, 128(4): 1229-1237.
- Chen X, Gong W, Shao X, Shi T, Zhang L, Dong J, Shi Y, Shen S, Qin J, Jiang Q: METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis 2022, 81(1): 87-99.
- Berenbaum F: Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage 2013, 21(1): 16-21.
- Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM, Crish JF, Bebek G, Ritter SY, Lindstrom TM et al: Identification of a central role for complement in osteoarthritis. Nat Med 2011, 17(12): 1674-1679.
- Zhang Y, Zhou S, Cai W, Han G, Li J, Chen M, Li H: Hypoxia/reoxygenation activates the JNK pathway and accelerates synovial senescence. Mol Med Rep 2020, 22(1): 265-276.
- Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T, Curran GL, Ogrodnik M, Jurk D, Johnson KO et al: Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J Gerontol A Biol Sci Med Sci 2017, 72(6): 780-785.
- Chou CH, Jain V, Gibson J, Attarian DE, Haraden CA, Yohn CB, Laberge RM, Gregory S, Kraus VB: Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci Rep 2020, 10(1): 10868.
- Sellam J, Berenbaum F: The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol 2010, 6(11): 625-635.
- Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H: Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 2011, 7(1): 33-42.
- Pearson MJ, Herndler-Brandstetter D, Tariq MA, Nicholson TA, Philp AM, Smith HL, Davis ET, Jones SW, Lord JM: IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci Rep 2017, 7(1): 3451.
- Aida Y, Maeno M, Suzuki N, Namba A, Motohashi M, Matsumoto M, Makimura M, Matsumura H: The effect of IL-1beta on the expression of inflammatory cytokines and their receptors in human chondrocytes. Life Sci 2006, 79(8): 764-771.
- Wang W, Li J, Li F, Peng J, Xu M, Shangguan Y, Li Y, Zhao Y, Qiu C, Qu R et al: Scutellarin suppresses cartilage destruction in osteoarthritis mouse model by inhibiting the NF-κB and PI3K/AKT signaling pathways. Int Immunopharmacol 2019, 77: 105928.
- Loeser RF, Goldring SR, Scanzello CR, Goldring MB: Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012, 64(6): 1697-1707.
- Primorac D, Molnar V: Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes (Basel) 2020, 11(8): 854.
- Sekelova T, Danisovic L: Rejuvenation of Senescent Mesenchymal Stem Cells to Prevent Age-Related Changes in Synovial Joints. Cell Transplant 2023, 32: 9636897231200065.
- Malaise O, Tachikart Y, Constantinides M, Mumme M, Ferreira-Lopez R, Noack S, Krettek C, Noël D, Wang J, Jorgensen C et al: Mesenchymal stem cell senescence alleviates their intrinsic and seno-suppressive paracrine properties contributing to osteoarthritis development. Aging (Albany NY) 2019, 11(20): 9128-9146.
- Miura Y, Endo K: Clearance of senescent cells with ABT-263 improves biological functions of synovial mesenchymal stem cells from osteoarthritis patients. Stem Cell Res Ther 2022, 13(1): 222.
- Santos Savio A, Machado Diaz AC, Chico Capote A, Miranda Navarro J, Rodríguez Alvarez Y, Bringas Pérez R, Estévez del Toro M, Guillen Nieto GE: Differential expression of pro-inflammatory cytokines IL-15Ralpha, IL-15, IL-6 and TNFalpha in synovial fluid from rheumatoid arthritis patients. BMC Musculoskelet Disord 2015, 16: 51.
- Liu W, Sun Y, He Y, Zhang H, Zheng Y, Yao Y, Zhang Z: IL-1β impedes the chondrogenic differentiation of synovial fluid mesenchymal stem cells in the human temporomandibular joint. Int J Mol Med 2017, 39(2): 317-326.
- Wei H, Shen G, Deng X, Lou D, Sun B, Wu H, Long L, Ding T, Zhao J: The role of IL-6 in bone marrow (BM)-derived mesenchymal stem cells (MSCs) proliferation and chondrogenesis. Cell Tissue Bank 2013, 14(4): 699-706.
- Fernandes TL, Kimura HA, Pinheiro CCG, Shimomura K, Nakamura N, Ferreira JR, Gomoll AH, Hernandez AJ, Bueno DF: Human Synovial Mesenchymal Stem Cells Good Manufacturing Practices for Articular Cartilage Regeneration. Tissue Eng Part C Methods 2018, 24(12): 709-716.
- Jeyaraman M, Muthu S: Synovium Derived Mesenchymal Stromal Cells (Sy-MSCs): A Promising Therapeutic Paradigm in the Management of Knee Osteoarthritis. Indian J Orthop 2022, 56(1): 1-15.
- Cao X, Wang X, Zhang W, Xia G, Zhang L, Wen Z, He J, Wang Z, Huang J, Wu S: WNT10A induces apoptosis of senescent synovial resident stem cells through Wnt/calcium pathway-mediated HDAC5 phosphorylation in OA joints. Bone 2021, 150: 116006.
- Kang C: Senolytics and Senostatics: A Two-Pronged Approach to Target Cellular Senescence for Delaying Aging and Age-Related Diseases. Mol Cells 2019, 42(12): 821-827.
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM: Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479(7372): 232-236.
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M et al: The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015, 14(4): 644-658.
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W et al: Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016, 22(1): 78-83.
- Sessions GA, Copp ME, Liu JY, Sinkler MA, D'Costa S, Diekman BO: Controlled induction and targeted elimination of p16(INK4a)-expressing chondrocytes in cartilage explant culture. Faseb j 2019, 33(11): 12364-12373.
- Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL et al: Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47: 446-456.
- Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N et al: Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40: 554-563.
- Keam SJ: Dasatinib: in chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. BioDrugs 2008, 22(1): 59-69.
- Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG: Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 2017, 23(9): 1072-1079.
- Elayyan J, Lee EJ, Gabay O, Smith CA, Qiq O, Reich E, Mobasheri A, Henrotin Y, Kimber SJ, Dvir-Ginzberg M: LEF1-mediated MMP13 gene expression is repressed by SIRT1 in human chondrocytes. FASEB J 2017, 31(7): 3116-3125.
- Li W, Cai L, Zhang Y, Cui L, Shen G: Intra-articular resveratrol injection prevents osteoarthritis progression in a mouse model by activating SIRT1 and thereby silencing HIF-2α. J Orthop Res 2015, 33(7): 1061-1070.
- Marouf BH, Hussain SA, Ali ZS, Ahmmad RS: Resveratrol Supplementation Reduces Pain and Inflammation in Knee Osteoarthritis Patients Treated with Meloxicam: A Randomized Placebo-Controlled Study. J Med Food 2018, https://doi.org/10.1089/jmf.2017.4176. Epub ahead of print.
- Yan S, Dong W, Li Z, Wei J, Han T, Wang J, Lin F: Metformin regulates chondrocyte senescence and proliferation through microRNA-34a/SIRT1 pathway in osteoarthritis. J Orthop Surg Res 2023, 18(1): 198.
- Chen J, Zhang J, Li J, Qin R, Lu N, Goltzman D, Miao D, Yang R: 1,25-Dihydroxyvitamin D Deficiency Accelerates Aging-related Osteoarthritis via Downregulation of Sirt1 in Mice. Int J Biol Sci 2023, 19(2): 610-624.
- Batshon G, Elayyan J, Qiq O, Reich E, Ben-Aderet L, Kandel L, Haze A, Steinmeyer J, Lefebvre V, Zhang H et al: Serum NT/CT SIRT1 ratio reflects early osteoarthritis and chondrosenescence. Ann Rheum Dis 2020, 79(10): 1370-1380.
- Matsuzaki T, Matsushita T, Takayama K, Matsumoto T, Nishida K, Kuroda R, Kurosaka M: Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann Rheum Dis 2014, 73(7): 1397-1404.
- Nishida K, Matsushita T, Takayama K, Tanaka T, Miyaji N, Ibaraki K, Araki D, Kanzaki N, Matsumoto T, Kuroda R: Intraperitoneal injection of the SIRT1 activator SRT1720 attenuates the progression of experimental osteoarthritis in mice. Bone Joint Res 2018, 7(3): 252-262.
- Miyaji N, Nishida K, Tanaka T, Araki D, Kanzaki N, Hoshino Y, Kuroda R, Matsushita T: Inhibition of Knee Osteoarthritis Progression in Mice by Administering SRT2014, an Activator of Silent Information Regulator 2 Ortholog 1. Cartilage 2021, 13(2_suppl): 1356s-1366s.
- Mercken EM, Mitchell SJ, Martin-Montalvo A, Minor RK, Almeida M, Gomes AP, Scheibye-Knudsen M, Palacios HH, Licata JJ, Zhang Y et al: SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 2014, 13(5): 787-796.
- Hoffmann E, Wald J, Lavu S, Roberts J, Beaumont C, Haddad J, Elliott P, Westphal C, Jacobson E: Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br J Clin Pharmacol 2013, 75(1): 186-196.
- Jiang C, Sun ZM, Hu JN, Jin Y, Guo Q, Xu JJ, Chen ZX, Jiang RH, Wu YS: Cyanidin ameliorates the progression of osteoarthritis via the Sirt6/NF-κB axis in vitro and in vivo. Food Funct 2019, 10(9): 5873-5885.
- Copp ME, Shine J, Brown HL, Nimmala KR, Hansen OB, Chubinskaya S, Collins JA, Loeser RF, Diekman BO: Sirtuin 6 activation rescues the age-related decline in DNA damage repair in primary human chondrocytes. Aging (Albany NY) 2023, 15(23): 13628-13645.
- Wang X, Li X, Zhou J, Lei Z, Yang X: Fisetin suppresses chondrocyte senescence and attenuates osteoarthritis progression by targeting sirtuin 6. Chem Biol Interact 2024, 390: 110890.
- Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL et al: Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36: 18-28.
- Zheng W, Feng Z, You S, Zhang H, Tao Z, Wang Q, Chen H, Wu Y: Fisetin inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes through activating SIRT1 and attenuates the progression of osteoarthritis in mice. Int Immunopharmacol 2017, 45: 135-147.
- Nogueira-Recalde U, Lorenzo-Gómez I, Blanco FJ, Loza MI, Grassi D, Shirinsky V, Shirinsky I, Lotz M, Robbins PD, Domínguez E et al: Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 2019, 45: 588-605.
- Zhang M, Mani SB, He Y, Hall AM, Xu L, Li Y, Zurakowski D, Jay GD, Warman ML: Induced superficial chondrocyte death reduces catabolic cartilage damage in murine posttraumatic osteoarthritis. J Clin Invest 2016, 126(8): 2893-2902.
- Yun MH: Cellular senescence in tissue repair: every cloud has a silver lining. Int J Dev Biol 2018, 62(6-7-8): 591-604.
- Yun MH, Davaapil H, Brockes JP: Recurrent turnover of senescent cells during regeneration of a complex structure. elife 2015, 4:e05505.
- Godwin JW, Pinto AR, Rosenthal NA: Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A 2013, 110(23): 9415-9420.
- Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N et al: NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest 2012, 122(7): 2601-2612.
- Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V et al: JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A 2015, 112(46): E6301-6310.
- Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW, Lee YS, Park SC: Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol 2017, 13(6): 616-623.
- Lee SJ, Jung YS, Yoon MH, Kang SM, Oh AY, Lee JH, Jun SY, Woo TG, Chun HY, Kim SK et al: Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J Clin Invest 2016, 126(10): 3879-3893.
- Bae YU, Choi JH, Nagy A, Sung HK, Kim JR: Antisenescence effect of mouse embryonic stem cell conditioned medium through a PDGF/FGF pathway. Faseb j 2016, 30(3): 1276-1286.
- Bae YU, Son Y, Kim CH, Kim KS, Hyun SH, Woo HG, Jee BA, Choi JH, Sung HK, Choi HC et al: Embryonic Stem Cell-Derived mmu-miR-291a-3p Inhibits Cellular Senescence in Human Dermal Fibroblasts Through the TGF-β Receptor 2 Pathway. J Gerontol A Biol Sci Med Sci 2019, 74(9): 1359-1367.
- Nasi S, Ea HK, So A, Busso N: Revisiting the Role of Interleukin-1 Pathway in Osteoarthritis: Interleukin-1α and -1β, and NLRP3 Inflammasome Are Not Involved in the Pathological Features of the Murine Menisectomy Model of Osteoarthritis. Front Pharmacol 2017, 8: 282.
- Dayer JM, Oliviero F, Punzi L: A Brief History of IL-1 and IL-1 Ra in Rheumatology. Front Pharmacol 2017, 8: 293.
- Kamath R, Simler G, Zhou C, Hart M, Joshi S, Ghayur T, Honore P: Blockade of both IL-1A and IL-1B by a combination of monoclonal antibodies prevents the development and reverses established pain in a preclinical model of osteoarthritis. Osteoarthritis Cartilage 2012, 20: S62.
- Fleischmann RM, Bliddal H, Blanco FJ: A Phase II Trial of Lutikizumab, an Anti-Interleukin-1α/β Dual Variable Domain Immunoglobulin, in Knee Osteoarthritis Patients With Synovitis. Arthritis Rheumatol 2019, 71(7): 1056-1069.
- Kloppenburg M, Ramonda R, Bobacz K, Kwok WY, Elewaut D, Huizinga TWJ, Kroon FPB: Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 2018, 77(12): 1757-1764.
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD et al: Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017, 377(12): 1119-1131.
- Schieker M, Conaghan PG: Effects of Interleukin-1β Inhibition on Incident Hip and Knee Replacement : Exploratory Analyses From a Randomized, Double-Blind, Placebo-Controlled Trial. Ann Intern Med 2020, 173(7): 509-515.
- Wu X, Cao L, Li F, Ma C, Liu G, Wang Q: Interleukin-6 from subchondral bone mesenchymal stem cells contributes to the pathological phenotypes of experimental osteoarthritis. Am J Transl Res 2018, 10(4): 1143-1154.
- Latourte A, Cherifi C, Maillet J, Ea HK, Bouaziz W, Funck-Brentano T, Cohen-Solal M, Hay E, Richette P: Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis 2017, 76(4): 748-755.
- de Hooge ASK, van de Loo FAJ, Bennink MB, Arntz OJ, de Hooge P, van den Berg WB: Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage 2005, 13(1): 66-73.
- Richette P, Latourte A: Efficacy of tocilizumab in patients with hand osteoarthritis: double blind, randomised, placebo-controlled, multicentre trial. Ann Rheum Dis 2021, 80(3): 349-355.
- Liu Y, Peng H, Meng Z, Wei M: Correlation of IL-17 Level in Synovia and Severity of Knee Osteoarthritis. Med Sci Monit 2015, 21: 1732-1736.
- Vincenti MP, Brinckerhoff CE: Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res 2002, 4(3): 157-164.
- Shiomi T, Lemaître V, D'Armiento J, Okada Y: Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol Int 2010, 60(7): 477-496.
- Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, Kokubun S, Bronner F: Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum 2005, 52(10): 3110-3124.
- Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T et al: Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest 2001, 107(1): 35-44.
- Wang M, Sampson ER, Jin H, Li J, Ke QH, Im HJ, Chen D: MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther 2013, 15(1): R5.
- Ni L, Lin Z, Hu S, Shi Y, Jiang Z, Zhao J, Zhou Y, Wu Y, Tian N, Sun L et al: Itaconate attenuates osteoarthritis by inhibiting STING/NF-κB axis in chondrocytes and promoting M2 polarization in macrophages. Biochem Pharmacol 2022, 198: 114935.
- Cai W, Zhang Y, Jin W, Wei S, Chen J, Zhong C, Zhong Y, Tu C, Peng H: Procyanidin B2 ameliorates the progression of osteoarthritis: An in vitro and in vivo study. Int Immunopharmacol 2022, 113(Pt A): 109336.
- Lou C, Deng A, Zheng H, Sun G, Zhao H, Li A, Liu Q, Li Y, Lv Z: Pinitol suppresses TNF-α-induced chondrocyte senescence. Cytokine 2020, 130: 155047.
- Kapoor N, Bhattacharjee A, Chakraborty S, Katti DS: Piperlongumine mediates amelioration of osteoarthritis via inhibition of chondrocyte senescence and inflammation in a goat ex vivo model. Eur J Pharmacol 2023, 961: 176136.
- Chen H, Tu M, Liu S: Dendrobine Alleviates Cellular Senescence and Osteoarthritis via the ROS/NF-κB Axis. Int J Mol Sci 2023, 24(3): 2365.
- Qin R, Sun J, Wu J, Chen L: Pyrroloquinoline quinone prevents knee osteoarthritis by inhibiting oxidative stress and chondrocyte senescence. Am J Transl Res 2019, 11(3): 1460-1472.
- Ji S, Xiong M, Chen H, Liu Y, Zhou L, Hong Y, Wang M, Wang C: Cellular rejuvenation: molecular mechanisms and potential therapeutic interventions for diseases. Signal Transduct Target Ther 2023, 8(1): 116.
- Jia Y, Qiu S, Xu J, Kang Q, Chai Y: Exosomes Secreted by Young Mesenchymal Stem Cells Promote New Bone Formation During Distraction Osteogenesis in Older Rats. Calcif Tissue Int 2020, 106(5): 509-517.
- Barrachina L, Remacha AR, Romero A, Vitoria A, Albareda J, Prades M, Roca M, Zaragoza P, Vázquez FJ, Rodellar C: Assessment of effectiveness and safety of repeat administration of proinflammatory primed allogeneic mesenchymal stem cells in an equine model of chemically induced osteoarthritis. BMC Vet Res 2018, 14(1): 241.
- Fu L, Hu Y, Song M, Liu Z, Zhang W, Yu FX, Wu J, Wang S, Izpisua Belmonte JC, Chan P: Up-regulation of FOXD1 by YAP alleviates senescence and osteoarthritis. PLoS Biol 2019, 17(4): e3000201.
- Ren X, Zhuang H: Ceria Nanoparticles Alleviated Osteoarthritis through Attenuating Senescence and Senescence-Associated Secretory Phenotype in Synoviocytes. Int J Mol Sci 2023, 24(5): 5056.
- Zhu J, Yang S: Stem cell-homing hydrogel-based miR-29b-5p delivery promotes cartilage regeneration by suppressing senescence in an osteoarthritis rat model. Sci Adv 2022, 8(13): eabk0011.
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dollé ME et al: An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014, 31(6): 722-733.
Asia-Pacific Journal of Pharmacotherapy & Toxicology
p-ISSN: 2788-6840
e-ISSN: 2788-6859
 Copyright © Asia Pac J Pharmacother Toxicol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Pharmacother Toxicol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.